Luck Lab
Research Publications Group Members BiographyIntegrative systems biology
Overview
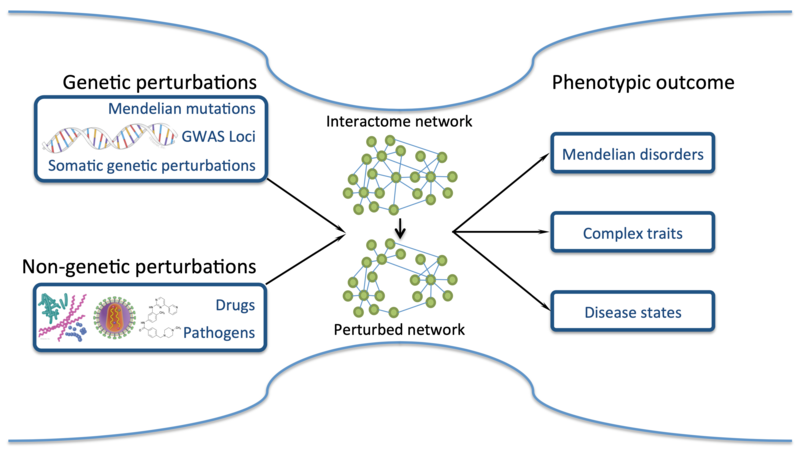
Genes do not function in isolation. Instead, their products, often proteins, engage in a multitude of physical and functional interactions with other molecules in the cell. These molecular interactions mediate all cellular functions and organisation. My lab is particularly interested in the dense ensemble of all interactions, referred to as the interactome, which is formed between proteins. Obtaining a comprehensive understanding of the protein interactome, both from a structural and functional point of view, is essential for understanding the molecular mechanisms that drive all cellular processes under physiological conditions, and for understanding how mutations, toxins and pathogens interfere with these processes and cause disease (Figure 1).
Conceptually, cells and other biological entities can be understood as systems defined by their components and the interactions between them. We understand that within a cell, all processes are connected, talk to each other and share components. Discovering this interconnectivity is difficult when focusing on one particular pathway or some of its components. We therefore employ a systems and omics data-driven approach to decipher the molecular mechanisms and principles of cellular function. We do so by combining computational and experimental approaches, enabling us to generate predictions and at the same time experimentally validate them. We invest in the development of integrative computational and experimental methods to efficiently annotate protein interactions with information on their structure, function and cellular context, and to interpret the effects of mutations on protein function. Primarily in collaborations, we apply our methods to processes such as chromatin remodelling, mRNA splicing, proteostasis and growth factor signalling, with a particular focus, where applicable, on deciphering the molecular mechanisms that drive phase separation. We aim to decipher the molecular mechanisms driving these processes under physiological conditions and study how mutations can alter them and cause neurodevelopmental disorders and age-related diseases such as neurodegeneration and cancer.
Prediction & experimental validation of interfaces in protein interactions
Identifying the regions and specific residues that mediate an interaction between two proteins is critical to predicting which mutations will perturb the interaction, which interaction partners have overlapping binding regions (and are therefore mutually exclusive), and which interactions can co-exist (and thus mediate the assembly of larger protein complexes). Furthermore, identifying binding regions often provides additional information on the molecular function of the interaction. However, most current assays that provide large amounts of protein interaction data do not provide any information on how proteins interact with each other (Figure 2). Downstream experimental characterisation of protein interaction interfaces often involves mutational scanning or protein fragmentation, which are both laborious and error-prone due to possible nonspecific perturbation of the entire protein fold. Many protein interactions are mediated by globular domains that either bind other types of globular domains or short stretches of amino acids (so-called linear motifs) in disordered regions of proteins. We develop novel approaches to predict known types of domain-domain and domain-linear motif interfaces using sequence-based methods and explore the use of artificial intelligence-based tools such as AlphaFold to discover novel interaction interface types. We implemented a medium-throughput experimental platform based on bioluminescence resonance energy transfer to validate interface predictions and test the effects of mutations on protein function.

Development of CL-MS for medium-throughput mapping of protein interaction interfaces
Cross-linking mass spectrometry (XL-MS) has emerged as a potentially powerful technique for mapping protein interactomes, deciphering protein complex topology and mapping protein interaction interfaces. However, it remains unclear which cross-linking reagents preferentially map to different types of protein interaction interfaces, whether some interaction interfaces are more or less amenable to detection by XL-MS, and whether protocols can be optimised to increase throughput. We build protein interaction reference datasets and are developing an experimental pipeline to benchmark and apply XL-MS for systematic characterisation of protein interaction interfaces.
Understanding the brain specificity of neurodevelopmental disorders
Neurodevelopmental disorders (NDDs) comprise various syndromes such as intellectual disability, developmental delay, autistic-like behaviour and epilepsy. Interestingly, more than 90% of the genes that have so far been linked to NDDs when mutated do not have a brain-specific expression pattern. This suggests that the brain-specific phenotypes caused by mutations in these proteins must result from perturbations within the context of their brain-specific network. We are integrating brain transcriptome and proteome data with protein interaction resources to predict the molecular mechanisms responsible for the brain-specific phenotypes of NDD-associated proteins that do not have brain-specific expression patterns. We will then test whether pathogenic but not benign mutations interfere with the predicted mechanisms in vitro and in vivo. We will focus in particular on proteins implicated in genome maintenance, in order to elucidate novel mechanisms that adapt genome maintenance processes to brain-specific contexts.
Deciphering protein-protein interaction interfaces that function in mRNA splicing
mRNA splicing is a key cellular process that massively extends the number of different protein forms in cells, enables additional layers of post-transcriptional regulation and is likely a key mechanism that drove the evolution of multicellularity. mRNA splicing itself is a highly regulated process involving dozens of proteins and RNA molecules that dynamically assemble as part of the spliceosome. Because of the dynamic nature and complexity of mRNA splicing, many steps are still poorly understood. In collaboration with the König lab at IMB and the Sattler group at the Technical University of Munich, we are using our computational and experimental techniques to elucidate the interfaces between proteins that function in mRNA splicing. We have discovered a novel core splice factor and determined its role in the splicing of particularly long introns (manuscript under revision).
Understanding molecular mechanisms of nuclear condensates that function in protein quality control
An increasing bulk of literature describes modes of protein-protein, protein-RNA and protein-DNA binding that drive molecular demixing in cells, leading to membraneless structures that display a continuum of liquid to solid states. While liquid condensates are associated with various advantageous properties for cellular processes such as sequestration of specific molecules and enhanced catalytic activity, solidification and aggregation of these assemblies are instead associated with pathogenic processes such as neurodegeneration and ageing. As part of the new Collaborative Research Centre (CRC) 1551 and in collaboration with the Beli lab at IMB and the Kukharenko and Kremer labs at the Max-Planck-Institute for Polymer Research in Mainz, we aim to elucidate the molecular mechanisms that drive the formation and resolution of nuclear proteasome-containing condensates and their role in proteostasis under conditions of proteotoxic stress.
Elucidating crosstalk between chromatin remodelling and genome maintenance
Various cellular processes such as replication, transcription, chromatin remodelling and DNA repair act simultaneously on the same DNA molecule. It is obvious that these processes need to be jointly orchestrated in order to not interfere with each other, but rather to act cooperatively to support each other’s function. As part of the CRC 1361 and in close collaboration with the Schick lab at IMB, we aim to elucidate the mechanisms by which chromatin remodelling complexes contribute to genome stability and DNA repair processes. We approach this problem with an integrative experimental and computational systems approach.
Funding
I am a fellow of the DFG’s Emmy Noether Program for young investigators since 2020, which has granted me €1.6m in funding to support my research ambitions for the coming 6 years. Further research projects are supported by the Alexander-von-Humboldt foundation, the CRC 1551 and the CRC 1361. IMB provides additional core funding.
Hiring
I am always open to receiving applications for internships, Bachelor and Master theses, PhD projects and postdoctoral research.